Saturs
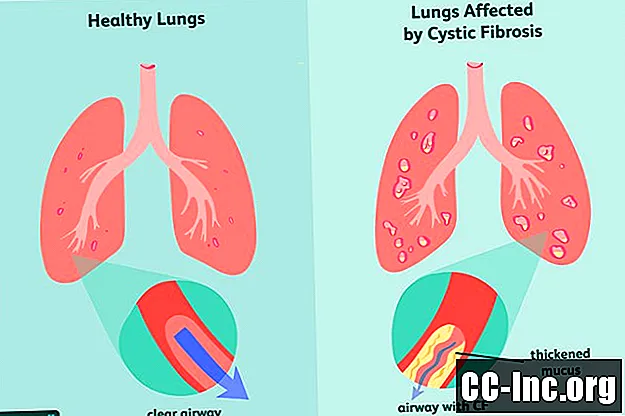
Cistiskā fibroze (CF) ir iedzimts, dzīvībai bīstams traucējums, kas bojā plaušas un gremošanas traktu. To izraisa bojāts gēns, kas izraisa sabiezējušu gļotu veidošanos, kas aizsprosto elpceļus un bloķē gremošanas enzīmu sekrēciju.Simptomi ir progresējoši un bieži izteikti, un tie var ietvert elpošanas problēmas, atkārtotas plaušu infekcijas, vāju augšanu, vīriešu neauglību un hronisku aizkuņģa dziedzera, aknu, nieru un sirds iekaisumu.
CF var diagnosticēt ar asins testiem, ģenētisko skrīningu un procedūru, kas pazīstama kā sviedru hlorīda tests.
Lai gan CF nav iespējams izārstēt, ir procedūras, kas var uzlabot gan cilvēka dzīves ilgumu, gan kvalitāti.
Tie ietver elpceļu attīrīšanas paņēmienus, inhalējamās antibiotikas, gļotu atšķaidītājus, aizkuņģa dziedzera fermentus, diētu ar augstu kaloriju daudzumu un jaunākas paaudzes medikamentus, kas pazīstami kā CFTR modulatori. Smagos gadījumos var būt nepieciešama plaušu transplantācija.
Cistiskās fibrozes simptomi
Kā ģenētisks traucējums cistiskā fibroze ir kaut kas, ar kuru jūs esat dzimis. Dzimšanas laikā tam var būt vai nav simptomu, un tas bieži var ilgt mēnešus vai pat gadus, pirms parādās jebkādas slimības pazīmes. Tajā laikā plaušās un gremošanas traktā, iespējams, jau ir bijuši bojājumi, kurus nevar atsaukt.
Visbiežāk sastopamās agrīnās CF pazīmes un simptomi ir:
- Zīdaiņa pirmā izkārnījuma (mekonija) bloķēšana
- Sāļās garšas āda
- Hronisks klepus, sēkšana vai krāsainas krēpas
- Brīvi, taukaini un parasti ar nepatīkamu smaku izkārnījumi
- Plaušu infekcija, bieži atkārtojas
- Slikta izaugsme un nespēja uzplaukt
Ja vien šos simptomus nevar kontrolēt, stresam uz plaušām (un nespējai iegūt svaru) var būt kumulatīvs efekts, kas ietekmē vairākus orgānus un palielina slimības komplikāciju risku.
Dažas no raksturīgākajām komplikācijām ir:
- Aizkavēta pubertāte
- Bronhektāzes (hroniska plaušu sieniņu sabiezēšana)
- Svara zudums
- Pankreatīts (aizkuņģa dziedzera iekaisums)
- Vīriešu neauglība
- Plaušu hipertensija (augsts asinsspiediens plaušās)
- Žultsakmeņi
- Ar cistisko fibrozi saistīts diabēts
- Cor pulmonale (labās puses sirds mazspēja)
- Ciroze (funkcionālas aknu rētas)
Tā kā CF izraisa progresējošu šūnu un audu ievainojumu, jebkurš plaušām un citiem orgāniem nodarīts kaitējums būs lielā mērā neatgriezenisks. Nāve visbiežāk būs elpošanas mazspējas rezultāts, kam seko sirds mazspēja un aknu mazspēja.
Cistiskās fibrozes simptomi
Cēloņi
Cistisko fibrozi izraisa cistiskās fibrozes transmembrānas receptoru (CFTR) gēna mutācija, kas ir atbildīga par CFTR olbaltumvielu ražošanu. Tas ir proteīns, kas organismam nepieciešams, lai regulētu sāls un ūdens plūsmu šūnās un ārpus tām . Ja olbaltumviela ir deformēta vai ar defektiem, tā var izraisīt dehidratāciju uz šūnas virsmas, izraisot apkārtējo gļotu sabiezēšanu.
CF ir autosomāli recesīvs traucējums, kas nozīmē, ka jums ir jāpārmanto CFTR mutācija gan no mātes, gan tēva, lai slimotu. Ja jūs mantojat tikai vienu bojātu gēnu, jums nebūs CF, bet tā vietā būsit mutācijas gēna nesējs.
Jūs varat mantot slimību, ja katram no jūsu vecākiem ir vai nu CFTR mutācija, vai arī pati CF. Ja abi vecāki ir pārvadātāji, jums ir:
- 25 procentu iespēja iegūt CF
- 50 procentu iespēja būt pārvadātājam
- 25 procentu iespēja tikt neietekmētai
No otras puses, ja viens no jūsu vecākiem ir pārvadātājs, bet otram ir CF, jums ir 50/50 izredzes vai nu iegūt CF, vai būt pārvadātājam.
Cistiskā fibroze ir viena no visbiežāk sastopamajām ģenētiskajām slimībām, kas skar apmēram vienu no katriem 2500 mazuļiem, kas dzimuši Amerikas Savienotajās Valstīs.
Tas ir visizplatītākais kaukāziešu un spāņu vidū, un retāk tas notiek Āfrikas vai Āzijas izcelsmes cilvēkiem.
Cistiskās fibrozes riska faktoriDiagnoze
Cistiskās fibrozes diagnosticēšanai tiek izmantoti daži testi. Viņi strādā, vai nu tieši atklājot CFTR mutāciju, vai netieši mērot bioloģiskās izmaiņas, kas atbilst slimībai. Diagnozes metode var atšķirties grūtniecības laikā, kad bērns piedzimst, vai jebkurā laikā pēc tam.
Cistiskās fibrozes ārstu diskusiju ceļvedis
Iegūstiet mūsu izdrukājamo ceļvedi nākamajai ārsta iecelšanai, lai palīdzētu jums uzdot pareizos jautājumus.

No diviem standarta testiem, ko parasti izmanto CF diagnosticēšanai:
- Sviedru hlorīda pārbaude, kas pazīstams arī vienkārši kā sviedru tests, mēra hlorīda daudzumu uz ādas. Tā kā CF traucē sāls pārnešanu uz šūnām un no tām, sviedros būs sāls uzkrāšanās.
- Ģenētiskā CFTR pārbaude lieto, lai noteiktu visbiežāk sastopamās CFTR mutācijas mutācijas. Kaut arī ir zināmas vairāk nekā 2000 CFTR mutācijas, kas izraisa cistisko fibrozi, 23, kas iekļautas standarta panelī, ir visticamāk aizdomās turamās personas.
Grūtniecības laikā CFTR ģenētisko testu var izmantot, lai pārbaudītu šķidrumus, kas iegūti, veicot amniocentēzi, vai šūnas, kas ekstrahētas, izmantojot koriona villus paraugus (CVS).
Jaundzimušo skrīnings arī parasti tiek izmantots CF diagnosticēšanai, un šodien tas ir pilnvarots visās 50 štatos un Kolumbijas apgabalā. Tas, ko tas nozīmē, atšķirsies atkarībā no tā, kur jūs dzīvojat Amerikas Savienotajās Valstīs. Ja jaundzimušo skrīninga rezultāti ir pozitīvi, diagnozes apstiprināšanai tiks izmantots sviedru tests.
Kā tiek diagnosticēta cistiskā fibrozeĀrstēšana
Kaut arī cistisko fibrozi nav iespējams izārstēt, ārstēšanas attīstība ir pagarinājusi to cilvēku dzīves ilgumu, kuri dzīvo ar šo slimību.
CF ārstēšanas mērķis ir četrkāršs: novērst infekcijas, saglabāt plaušu darbību, normalizēt gremošanu un palēnināt slimības progresēšanu.
Starp terapeitiskajiem instrumentiem, ko izmanto CF pārvaldīšanai:
- Elpceļu attīrīšanas paņēmieni (ACT) tiek veikti, lai izvadītu un izvadītu uzkrāto gļotu no plaušām. Paņēmieni ietver klepus klepu, krūšu krūtis vai krūšu sienas svārstības.
- Diēta ar augstu tauku saturu un augstu kaloriju daudzumu lieto, lai kompensētu tauku, olbaltumvielu un barības vielu absorbcijas traucējumus zarnās.
- Aizkuņģa dziedzera enzīmu piedevas tiek izmantoti gremošanas enzīmu stiprināšanai, kurus aizkuņģa dziedzeris nevar radīt pārmērīgas gļotu uzkrāšanās dēļ.
- Antibiotikas lieto katru dienu, lai novērstu bakteriālas plaušu infekcijas.
- Mukolītiskie līdzekļi- drīkst lietot zāles, kas pirms ACTs izmantotas gļotu novājēšanai.
- CFTR modulatori ir jauna zāļu klase, kas var novērst dažus CFTR olbaltumvielu defektus un atjaunot to regulējošo funkciju.
- Skābekļa terapija var lietot akūtu epizožu laikā, kad elpošana ir nopietni traucēta.
- Enterālais uzturs, kas pazīstams arī kā barošana caurulēs, var izmantot, ja jūs nevarat uzturēt svaru ar normālu uzturu.
- Plaušu transplantācija tiek uzskatīts, kad jūsu plaušas vairs nevar atbalstīt izdzīvošanu bez mehāniskas ventilācijas.
Tikt galā
1938. gadā, kad cistiskā fibroze pirmo reizi tika klasificēta kā slimība, bērni reti nodzīvoja pēc pirmā dzīves gada. Līdz astoņdesmitajiem gadiem varēja sagaidīt, ka viņš nodzīvos 20 līdz 25 gadus. Mūsdienās attēls ir pilnībā mainījies ar cilvēkiem, kuri dzīvo labi vecumā no 40 līdz pat 50 gadiem, ja ārstēšana tiek uzsākta agri un tiek ievērota.
Tas nenozīmē, ka CF būtu mazāk nopietns nekā jebkad agrāk. Tas ir notikums, kas maina dzīvi, un tam nepieciešama rūpība un konsekvence, lai ne tikai tiktu galā ar šo slimību, bet arī dzīvotu pēc iespējas augstāku dzīves līmeni.
Šajā nolūkā jums jā normalizē CF savā dzīvē, izveidojot kārtību un praksi, lai izvairītos no kāpumiem un kritumiem, kas var izraisīt stresu un palielināt invaliditāti. Starp apsvērumiem jums būs nepieciešams:
- Pārvaldiet savu uzturu. Cilvēkiem ar CF bieži vien ir vajadzīgas divreiz vairāk kaloriju nekā dienā.
- Regulāri vingrojiet. Ideālā gadījumā fitnesa rutīnā vajadzētu ietvert vismaz 20 līdz 30 minūtes ilgu aerobo aktivitāti trīs reizes nedēļā. Atrodiet kaut ko patīkamu, ko varat darīt visu mūžu.
- Uzturiet labi mitrinātu. Šādi rīkojoties, plaušas un zarnas darbojas pareizi. Atkarībā no vecuma jums vajadzētu dzert ne mazāk kā sešas līdz astoņas augstas glāzes ūdens dienā.
- Pareizi veiciet elpceļu klīrensu. Mainoties jūsu veselības vajadzībām, var mainīties arī nepieciešamo klīrensa rīku veidi. Runājiet ar pulmonologu vai fizioterapeitu, ja nesasniedzat nepieciešamos rezultātus.
- Meklējiet atbalstu. Papildus draugiem un ģimenei varat sazināties ar tuvāko Cistiskās fibrozes fonda (CFF) nodaļu, lai pievienotos atbalsta tīklam savā apkārtnē.
- Meklējiet finansiālu palīdzību. CFF piedāvā pakalpojumus, kas palīdz ģimenēm labāk tikt galā ar augstajām CF ārstēšanas izmaksām.
Vārds no Verywell
Kaut arī jaundzimušo skrīnings ir dramatiski palielinājis CF diagnozes līmeni zīdaiņiem, vairāk nekā 25 procenti diagnožu tiek veikti tikai bērnībā, pusaudžos un agrā pieaugušā vecumā.
Tas ir problemātiski, jo agrīna diagnostika un ārstēšana var novērst daudzas smagākas CF komplikācijas, pirms var tikt nodarīts nopietns kaitējums. Kaut arī ārstēšana nevar apturēt vai mainīt slimību, tā var nodrošināt daudz vairāk gadu bez slimībām.
Šim nolūkam ir svarīgi zināt agrīnos CF simptomus un runāt ar savu ārstu, ja jums ir aizdomas, ka bērnam varētu būt šī slimība. Tas jo īpaši attiecas uz valstīm, kurās ekrāns tiek veikts tikai ar IRT asins analīzēm, kā rezultātā 5% bērnu var piedzīvot novēlotu diagnozi vai kļūdaini negatīvu rezultātu, saskaņā ar Viskonsinas Universitātes Medicīnas un sabiedrības veselības skolas pētījumu .
Kādus simptomus jūs varat sagaidīt ar cistisko fibrozi?